About
PhD in cheminformatics. I am interested in applying machine learning, especially deep learning, techniques for solving challenging problems in chemistry, drug discovery and environmental toxicology.
My primary research interests involve:
- Quantitative Structure-Activity Relationships (QSAR) modeling.
- Development of data-driven and interpretable molecular representations via deep learning.
- Molecular informatics approach for visualizing and navigating chemical space.
Research Projects
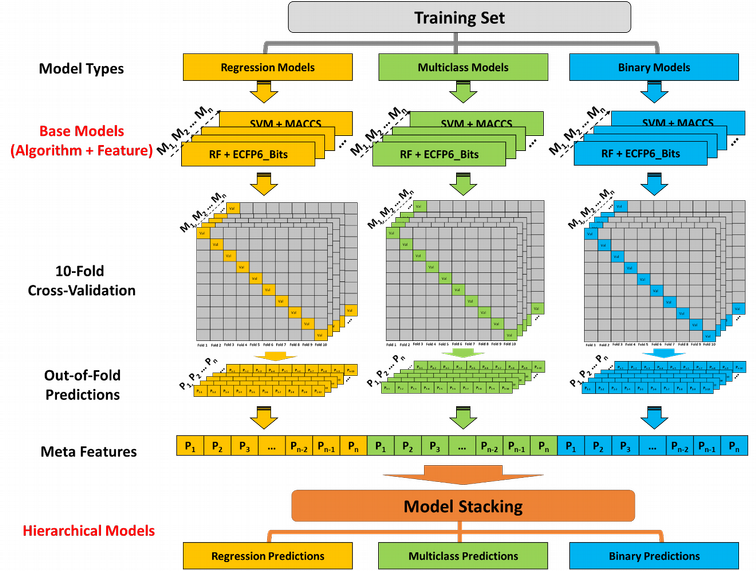
Reliable in silico approaches to replace animal testing for the evaluation of potential acute toxic effects are highly demanded by regulatory agencies. The first research project of my Ph.D. was developing a hierarchical QSAR modeling method that integrates binary, multiclass and regression models for predicting three acute oral systemic toxicity endpoints used by a variety of regulatory bodies for toxicity test. This method represents (1) a promising alternative prediction method to animal testing; (2) a more efficient and powerful ensemble method compared to the model consensus (predictions averaging). This project is a collaboration with Dr. Nicole Kleinstreuer at the NIEHS and published on Chemical Research in Toxicology. Github
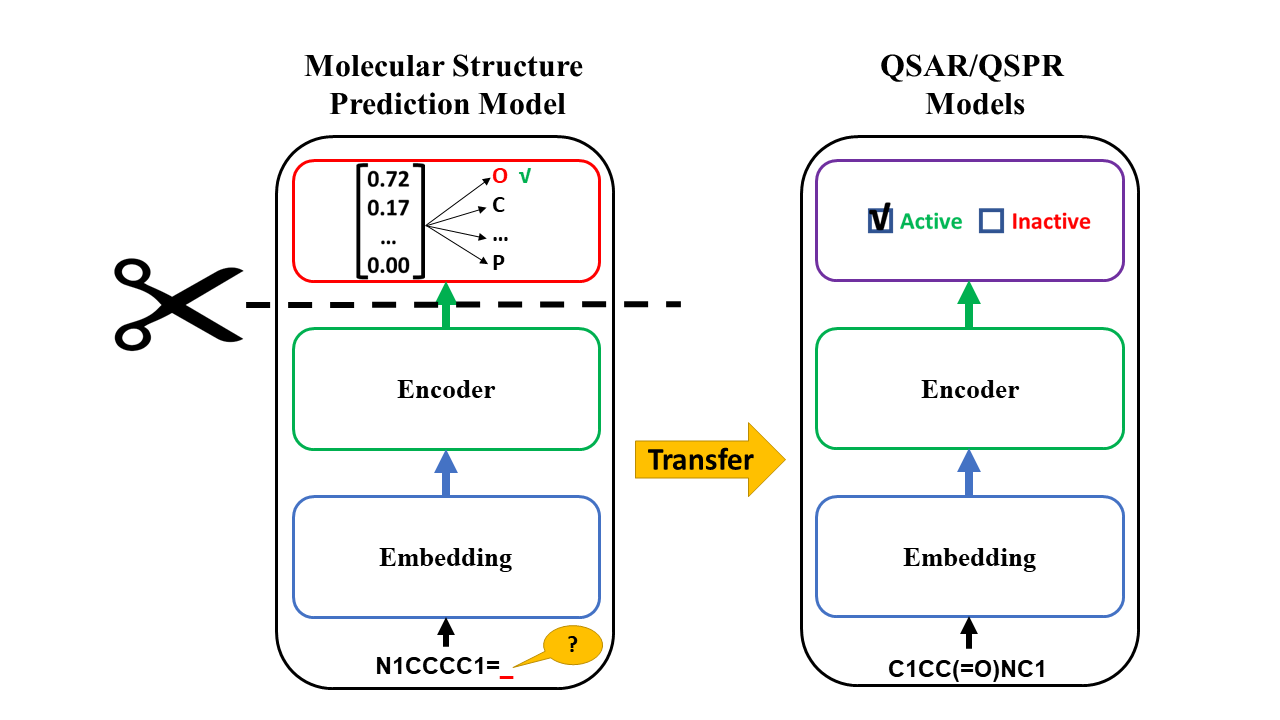
Quantitative Structure–Activity Relationships (QSAR) are statistical, data-driven models that establish quantitative links between an experimental activity (e.g., binding affinity, inhibition potency) and chemical structures. QSAR models are typically developed using supervised machine learning algorithms and further validated using a variety of statistical procedures and metrics. Good model performance usually requires a decent amount of labeled data, but collecting labels is expensive and hard to be scaled up. Thus, it would be highly relevant to utilize the tremendous unlabeled compounds from publicly-available datasets. Self-supervised learning opens up a huge opportunity for better utilizing unlabeled data. Molecular Prediction Model Fine-Tuning (MolPMoFiT) approach, an effective transfer learning method based on self-supervised pre-training + task-specific fine-tuning for QSAR modeling (Journal of Cheminformatics ). Github
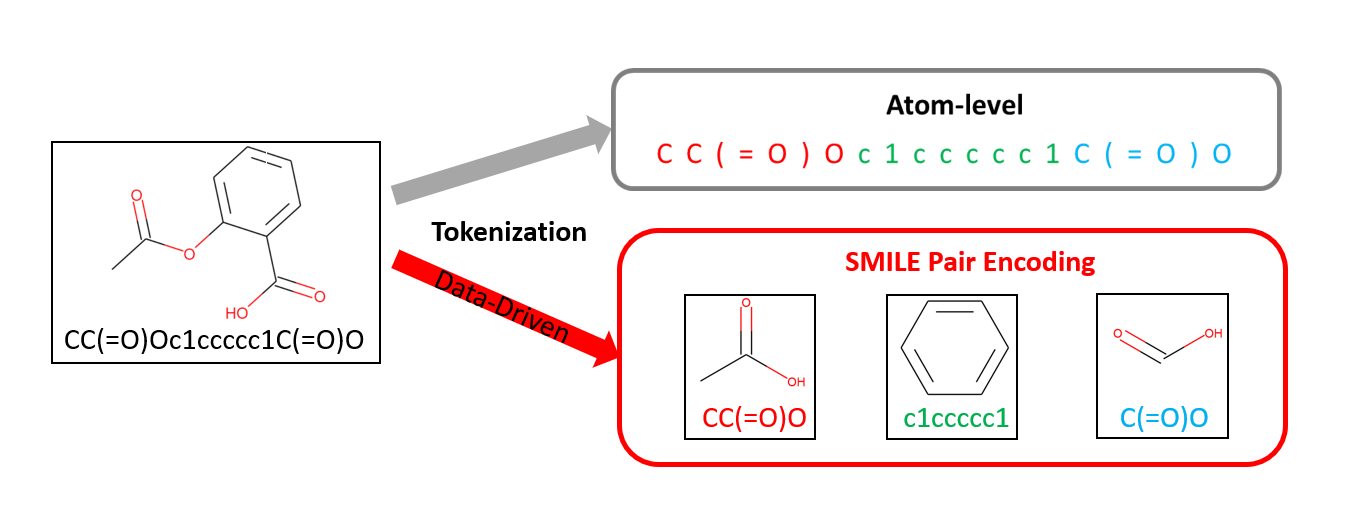
SMILES Pair Encoding (SPE) is a data-driven substructure tokenization algorithm for deep learning. SPE learns a vocabulary of high frequency SMILES substrings from ChEMBL and then tokenizes new SMILES into a sequence of tokens for deep learning models. SPE splits SMILES into human-readable and chemically explainable substrings and shows superior performances on both generative and predictive tasks compared to the atom-level tokenization. [ChemRxiv] [Github]